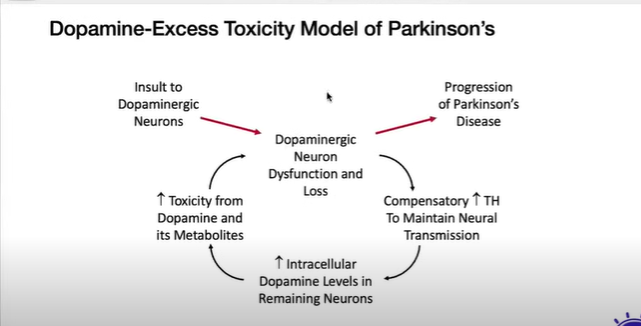
Dr. Jonathan Sackner-Bernstein presents evidence that the standard treatment to alleviate symptoms of Parkinson’s Disease (L-dopa) may be doing more harm than good in the long run. According to the standard view, deaths of these dopaminergic neurons leads to insufficient inhibition of motor neurons, resulting in Parkinsonian symptoms. Giving L-dopa combats these symptoms: L-dopa is absorbed by the remaining neurons and converted to dopamine, thus increasing the amount released per neuron at the synapses and thereby helping to restore inhibition. However, data indicate that the remaining neurons actually increase their synthesis of dopamine in response to the loss of neurons, i.e., loss of inhibition leads to increased synthesis in a negative feedback loop. However, for some reasons much of this dopamine enters the cell’s synaptic protoplasm instead of being packaged into vesicles as it normally would be. There it acts as a lethal toxin that eventually kills the cell.
If this view is correct, then the traditional treatment with L-dopa is actually accelerating the deaths of remaining dopaminergic neurons despite its ability to ameliorate symptoms in the short run. A new treatment has been proposed that treats the disorder by blocking the toxicity of dopamine within the neuron. “Why I am optimistic that we might now have a Silver Bullet for PD”
This is really interesting stuff. Though being a bear of little brain it’s pretty hard for me to understand. But there sure seem to be some interesting control processes – or, more specifically, the failure thereof – involved in Parkinson’s.
I haven’t listen to the whole lecture but I did get to what Bernstein calls his “model” of Parkinson’s:
I think this model is describing a control process but I have a couple questions about how it could be turned into a working model.
The controlled variable here seems to be “neural transmission”. If that’s true, how is that sensed by the cell or cell group? Is it maintained at a fixed reference level?
Besides insults to the dopaminergic neurons (aren’t all neurons dopaminergic?) it seems that there must be other disturbances to dopamine levels that affect neural transmission since the TH enzyme “compensation” system is always in effect. Perhaps neural transmission itself affects dopamine levels?
Could you try to sketch out the beginnings of a PCT type control model of neural transmission control – or whatever it is that is controlled – and the effect of Parkinson’s disease on that process. I think these medical people know a lot about the relationships between the variables involved in dopamine production and neural transmission but I think with a good control model of how these relationships actually result in control they could make even greater progress towards understanding Parkinson’s and how it might actually be cured.
I’m glad you’re looking at this, Rick. I’ve got too much going on just now to take it up.
You might get more out of
Sackner-Bernstein, J. Rethinking Parkinson’s disease: could dopamine reduction therapy have clinical utility?. J Neurol (2024). https://doi.org/10.1007/s00415-024-12526-7
No. Search for ‘ergic’ in the Wikipedia article about neurotransmitters to see something of the variety. Operative questions are where are receptors for a given neurotransmitter, and what is the effect e.g. on a synapse with an X-ergic receptor when it receives molecules of X neurotransmitter. This is the ambient ‘soups’ aspect of living control systems in the now resolved ‘soups vs. sparks’ controversy. (It’s both, analogous to the resolution that light is both photons and waves.) See The War of the Soups and the Sparks.
Mogi, M., Harada, M., Kiuchi, K. et al. Homospecific activity (activity per enzyme protein) of tyrosine hydroxylase increases in parkinsonian brain. J. Neural Transmission 72, 77–82 (1988). https://doi.org/10.1007/BF01244634
“Neural transmission” is a perceptual variable in the outside human observer. It cannot be a variable perceived and controlled by the neuron or Bill’s ‘nerve fiber’ (parallel bundle of dendrites or axons). If a neuron could perceive and control its rate of firing it would conflict with control of that rate by the control loop of which that neuron is a segment.
“Neurotransmitters are released into and diffuse across the synaptic cleft, where they bind to specific receptors on the membrane of the postsynaptic neuron. … Depending on the receptor, binding of neurotransmitters may cause excitation, inhibition, or modulation of the postsynaptic neuron.” (From that WP article on neurotransmitters.)
Excitation, inhibition, and modulation are effects on what is observed as neural transmission, effects which are separate from input up the hierarchy from environmental effects on sensor cells.
Thanks Bruce. This was very helpful. I have a few questions some of which may have been answered in the Sackner-Bernstein video but I didn’t get them.
Do dopaminergic neurons group together in a nerve? It seems that they do based on Bruce A.'a description of the process: “…data indicate that the remaining neurons actually increase their synthesis of dopamine in response to the loss of neurons, i.e., loss of inhibition leads to increased synthesis in a negative feedback loop.”
How do the remaining neurons know that they should increase their synthesis of dopamine when there is loss of inhibition? Inhibition presumably results from loss (or lowered) of transmission of dopamine into the synaptic cleft. Can the dopaminergic neuron sense the concentration of dopamine released into the synaptic cleft or does it actually sense the level of inhibition produced in the dendrite receptors? Again, it’s a question of what variable is being controlled by the negative feedback loop. My guess is that it must by the concentration of dopamine.
Apparently when surviving dopaminergic neurons increase their synthesis of dopamine to compensate for the loss of other dopaminergic neurons, they start making so much of it that some proportion of the dopamine can’t be packed into vesicles where it would be useful for synaptic transmission, but instead it remains in the cell’s protoplasm where it is turned into a poisonous substance that eventually kills the cell. Is this right? If so, it implies that the density of dopamine in the cell’s protoplasm is not controlled, which would be strange since, under normal circumstances, the cell is presumably continuously synthesizing dopamine and there would be a dangerous buildup of dopamine in the protoplasm if the neuron didn’t dissipate it at the appropriate rate via the vesicles dumping dopamine into the synaptic cleft. If this description of the situation is correct, then my guess is that dopamine density in the protoplasm is a controlled variable in a one-way control system, which increases dopamine synthesis when the level is below the reference but can’t do anything about it – can’t destroy the excess protoplasmic dopamine – when dopamine density is above the reference.
Dopamine is not transmitted in a targeted sense, it’s in greater or lesser abundance in the intercellular environment. Inhibition is an active process, but I don’t know the mechanism. That Wikipedia page is a good start. It may involve reuptake, by which dopamine is reabsorbed from circulation. I see that norepinephrine neurons may be involved in uptake of dopamine. Dopamine reuptake inhibitors are an important class of psychiatric drugs.